 Run HyPare Run HyPare |
Features |
Theory |
|
| Algorithm |
Use Scenarios |
References |
|
| View an example |
Introduction
HyPare is a newly designed tool based on the PARE algorithm (Predicting Association Rate Enhancement).PARE calculates the association rate (kon) of a mutant complex from the Debye Huckle energy of interaction (charge charge interactions) of the two proteins and from an experimentally measured kon value.
PARE prediction power was validated with the complexes barnase/barstar, tem/blip, hirudin/thrombin, and acetylcholinesterase/fasciculin.
Using the PARE platform, HyPare was designed to allow the processing of multiple protein complexes, with newly added features.
Among others, HyPare explores a protein's sequence, introducing charge mutation at each residual position, and thus providing a powerful method of detecting hotspots for association (hotspots are residue positions that significantly alter the association rate upon mutation).
Features...
- Design of faster and tighter binding proteins.
Detection of hotspots for association.
Analysis of hotspots according to interface regions.
Prediction of the rate of association at different salt concentrations.
Robustness: HyPare can handle many typical errors in pdb files.
Direct access to the Protein Database (PDB).
Multiple complexes processing (batch work).
Graphical representation of your results.
Visual perception of your output with Jmol applet.
One click export of your output into easily handled tab delimited tables.
Saving rate increase potential of each residue point into the bfactor section of
a newly created pdb file.
Use Scenarios...
HyPare was used in the analysis of protein protein association for many different
complexes and for different purposes. Here are some of the use cases where HyPare was successfully applied:
- Mutant complex kon prediction: Given the rate of association and the coordinates of a wild type complex, and the coordinates of a mutant complex, HyPare can predict the rate of association for the mutant one.Hotspots prediction: Given the coordinates of a wild type complex, HyPare scans the entire sequence of each protein partner and predicts which residue should be charge mutated in order to increase or decrease the rate of association.Interaction detection: Giving a set of interacting chains, HyPare can calculate the Coulombic energy of interaction of each pair, providing a method to rank the interactions.Docking filtration: Docking algorithms usually yield thousands of docked solutions. One major challenge is to narrow down this large set of solutions into a much smaller one. HyPare satisfies this challenge and provides an electrostatic based filter. The analysis of the docking solutions can be further refined with our interface predictor ProMate, which can be used to score the docking resultsIonic strength: The rate of association is often measured with a particular salt concentration. HyPare can calculate the new rate of association under different salt concentrations at which kon was not measured experimentally.
Short background...
The rate of association is a function of an electrostatic independent basal rate that is fixed for a given complex, and an electrostatic dependent component that is a function of the Coulombic energy of interaction -DU/RT (DU is the electrostatic energy of interaction. R is the gas constant and T is the temperature). Combining these two gives:
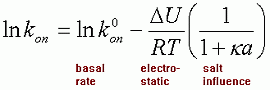 |
(1) |
where a is set to 6A, and k is the Debye-huckel screening parameter that
relates to the ionic strength of the solution.
U is calculated using:
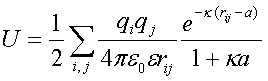 |
(2) |
where i and j are the charged atoms in the proteins and e is the
dielectric constant.
The electrostatic energy of interaction is calculated
from the difference between the electrostatic energy of the complex and the electrostatic energy of the two individual proteins.
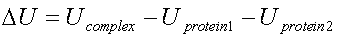 |
(3) |
where Ucomplex and Uprotein are calculated from equation 2.
The relation between kon and DU at an ionic strength of 0.022M is:
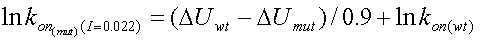 |
(4) |
where lnkon(wt) has to be determined experimentally. The adjustment of the rate to the rate at I=0.022 is done automatically by the program according to:
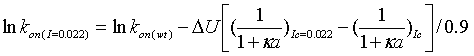 |
(5) |
Calculating kon at any other salt concentration can be done using the formula:
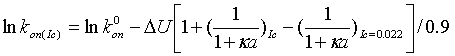 |
(6) |
where lnk0on, the basal rate, is found using:
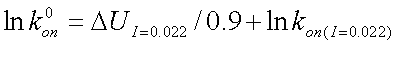 |
(7) |
where DU is calculated from equation 2 and lnkon(I=0.022) is calculated from equation 5.
The energy calculation is performed using the following parameters:
- A fixed dielectric constant set
to 80
The ionic strength of the solution is set
to 0.022M
The program assigns charges only to atoms
of charged residues (ARG +1, LYS +1, ASP -1, GLU -1, C terminus (OXT) -1, N
terminus +1, and charged heteroatoms) Each full charge is distributed over one or two of the
atoms in the residue. Click here to view the complete charge rules.
References...
Theoretical background and experimental evidence for the tool can be found at:
- Yossi Shaul and Gideon Schreiber (2004). Exploring the charge space of protein-protein association: A proteomic
study. Submitted.
- C. Kiel, T. Selzer, Y. Shaul, G. Schreiber, and C. Herrmann (2004). Electrostatically optimized Ras-binding Ral guanine dissociation stimulator mutants increase the rate of association by stabilizing the encounter complex. PNAS 2004 101: 9223-9228. PMID: 15197281
- Selzer, T. and Schreiber, G. (2001). New Insights Into the Mechanism of Protein-Protein Association. Proteins. 45, 190-198. PMID: 11599022
- Selzer, T., Albeck, S., and Schreiber, G.,(2000). Rational Design of Faster Associating and Tighter Binding Protein Complexes. Nat. Struct. Biol. 7, 537-541. PMID: 10876236
- Selzer, T. and Schreiber, G. (1999). Predicting the Rate Enhancement of Protein Complex Formation from the Electrostatic Energy of Interaction. J. Mol. Biol. 287, 409-419. PMID: 10080902