Input Options
- pdb id(s): Enter your pdb id code and the chain id's. For example,
A B 1jtg. Alternatively, you may upload your own coordinates files.
NOTE: Any atom that is not assigned a chain ID will be ignored.
This is often the case with heteroatoms. Therefore, if you want
them to be included in the calculation you must edit the file to
include the chain IDs.
- Running conditions:
- first PDB entry is wild-type and rest are mutants: When checked, HyPare assumes that the first pdb entry is wt and the rest of the entries are mutants. In that case, the basal rate of the wt complex will be used to calcuate the association rate of the mutant complexes as well. When unchecked (default), HyPare treats each complex independently, each with its own basal rate.
- calculate energy and find hotspots: When this option is selected (default), HyPare will calculate the Coulombic energy for each complex and will find hotspots for every chain (Hotspots are those residues that significantly alter the association rate upon charge mutations). This option is recommended in the design of faster and tighter binding proteins.
- calculate energy only: When selected, HyPare will calculate the Coulombic energy for each complex, and will not attmept to find hotspots. This option is recommended if you are interested in the energy value only, e.g. as an electrostatic filter in docking applications.
- first PDB entry is wild-type and rest are mutants: When checked, HyPare assumes that the first pdb entry is wt and the rest of the entries are mutants. In that case, the basal rate of the wt complex will be used to calcuate the association rate of the mutant complexes as well. When unchecked (default), HyPare treats each complex independently, each with its own basal rate.
- Experimental Ionic Strength: You must enter this value in order
to calculate the predicted kon value at the target strength.
The ionic Strength is calculated as follows:

This is an important parameter that corrects the concentration (c) for differences in ion valences (z). In calculating the ionic strength we recommend that you include all ion sources, as from salt and buffers. NOTE: if you are only interested in finding hotspots, you can leave this field out
- Target Ionic Strength: Here you enter the ionic strength at which you want
the new kon to be calculated at, and at which you want hotspots to be calculated.
- Association rate( kon): This is the association rate as measured experimentally.
If you are not sure what is your kon value, HyPare can still find hotspots,
as the calculation depends on a ratio of rates. However, if want to know the exact kon
at the target salt, you must enter this value
- Charge Rules: You may choose from 3 options:
- Use default charge rules: This is the recommended option. click here
to view these rules
- Append my own charge rules: If you like the default rules but you wish to add or modify a an existing entry, then use
this option. Please note, you must upload your entries in a text file. If one or more of
your rules contradicts with the default rules, then your rules are granted priority.
- Use my own charge rules only: Use this last option if you would like to upload a completely different set of rules In that case, only your rules will be used, and the default ones will be discarded
- Use default charge rules: This is the recommended option. click here
to view these rules
Equations Used
The rate of association is a function of an electrostatic independent basal rate that is fixed for a given complex, and an electrostatic dependent component that is a function of the Coulombic energy of interaction -DU/RT (DU is the electrostatic energy of interaction. R is the gas constant and T is the temperature). Combining these two gives:
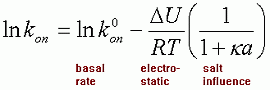 |
(1) |
where a is set to 6A, and k is the Debye-huckel screening parameter that
relates to the ionic strength of the solution.
U is calculated using:
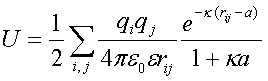 |
(2) |
where i and j are the charged atoms in the proteins and e is the
dielectric constant.
The electrostatic energy of interaction is calculated
from the difference between the electrostatic energy of the complex and the electrostatic energy of the two individual proteins.
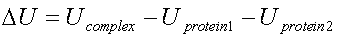 |
(3) |
where Ucomplex and Uprotein are calculated from equation 2.
The relation between kon and DU at an ionic strength of 0.022M is:
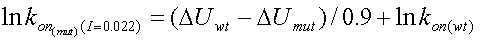 |
(4) |
where lnkon(wt) has to be determined experimentally.
The adjustment of the rate to the rate at I=0.022 is done automatically by the program according to:
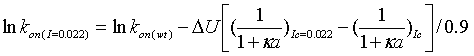 |
(5) |
Calculating kon at any other salt concentration can be done using the formula:
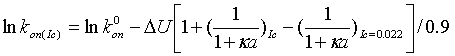 |
(6) |
where lnk0on, the basal rate, is found using:
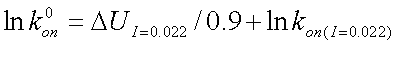 |
(7) |
where DU is calculated from equation 2 and lnkon(I=0.022) is calculated from equation 5.
Charge Rules List
- The Program assigns charges to selected atoms as mentioned below.
- The N terminus is automatically assigned a charge of +1.
- The C terminus is assigned a charge of -1 if an OXT entry is found in the pdb.
- Atoms of neutral residues that are assigned a zero charge are modified in the hotspots finding process. Therefore, its important to include these atoms, although they carry a charge of zero, because these are the hypothetical sites for charge mutations.
- The list below is the default charge rules. However, you may upload your list, as long as you follow the format: ATOM_NAME RESIDUE CHARGE, where each charge rule entry is separated by a newline and each column is separated by space or tab.
For example:
NH1 ARG 0.5
NH2 ARG 0.5
CA HIS 1.0
|
|